
Multiple Sequence Alignment; Part One
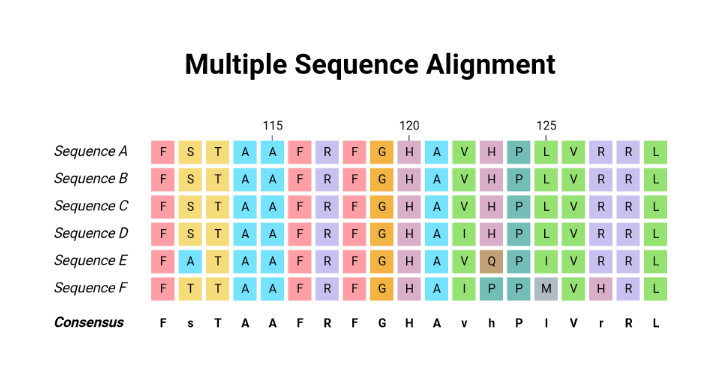
Multiple sequence alignment (MSA) is a computational technique that aligns three or more biological sequences, typically DNA, RNA, or proteins. The aim is to arrange these sequences in a way that highlights their homologies and differences, revealing regions of shared ancestry and potential functional importance. The complexity of MSA increases with higher numbers of the sequences being aligned.
MSA methods
There are multiple algorithms used for MSA, each with its strengths and weaknesses. Popular methods include:
- Progressive Alignment P-MSA: Sequences are aligned pairwise, step by step, gradually building a larger alignment, according to a pre-ordered sequence.
- Iterative Refinement: An initial alignment is created, then refined and realigned based on scoring functions that reward matches and penalize mismatches, filling the gaps of P-MSA.
- Structure-based Alignment: Aligns sequences based on the predicted 3D structures of the proteins they encode.
These algorithms use scoring matrices that assign points for matches, mismatches, and gaps (insertions/deletions). The goal is to maximize the score, reflecting the optimal alignment with minimal differences.
MSA most common software programs:
- Clustal Omega: A widely used and versatile tool for both protein and nucleotide sequence alignment. It offers various algorithms and options for fine-tuning alignments.
- Muscle: known for its speed and accuracy for protein alignments. It offers different alignment modes and supports large datasets.
- MAFFT: Highly customizable software with various algorithms and options for both protein and nucleotide alignments. It excels at handling large datasets and is known for its speed.
- T-Coffee: Offers multiple alignment algorithms and visualization tools. It is particularly useful for aligning distantly related sequences.
This following video from Jalview, a free software alignment software, will guide you on your MSA process.
Choosing the Right Software:
The best software for you will depend on your specific research needs and preferences. Some factors to consider include:
- The type of sequences you are working with (protein, nucleotide)
- The size of your datasets
- The desired level of accuracy and speed
- The available features and functionalities
- Your budget
Applications of MSA:
MSA is a cornerstone of various biological disciplines:
- Evolutionary studies: By analyzing conserved regions in MSAs, we can infer evolutionary relationships between organisms and reconstruct their ancestral sequences.
- Function prediction: Identifying conserved residues in functional domains within proteins can help predict their function in unknown sequences.
- Drug discovery: Comparing proteins from different organisms can help identify potential drug targets based on conserved functional regions.
- Structure prediction: MSA can be used to predict the 3D structure of a protein based on the structures of related proteins with known structures.
Conclusion:
No single MSA algorithm is perfect. The accuracy of alignments depends on various factors, including sequence similarity, algorithm choice, and parameter settings. Additionally, interpreting gaps and insertions introduced during alignment can be challenging.
Nevertheless, MSA is a powerful tool that explains shared history and functional potential of biological sequences. As sequencing technologies advance and algorithms are refined, this technique will continue to unlock new secrets from the vast libraries of life's code. So, the next time you see a complex protein alignment, remember the detective work and hidden stories it reveals!